ο Θεραπεία αντικατάστασης ενζύμου χρησιμοποιείται για τη θεραπεία λυσοσωμικών ασθενειών αποθήκευσης, στις οποίες η έλλειψη ενζύμων οδηγεί σε παθολογική συσσώρευση προϊόντων αποδόμησης στα λυσοσώματα των κυττάρων.
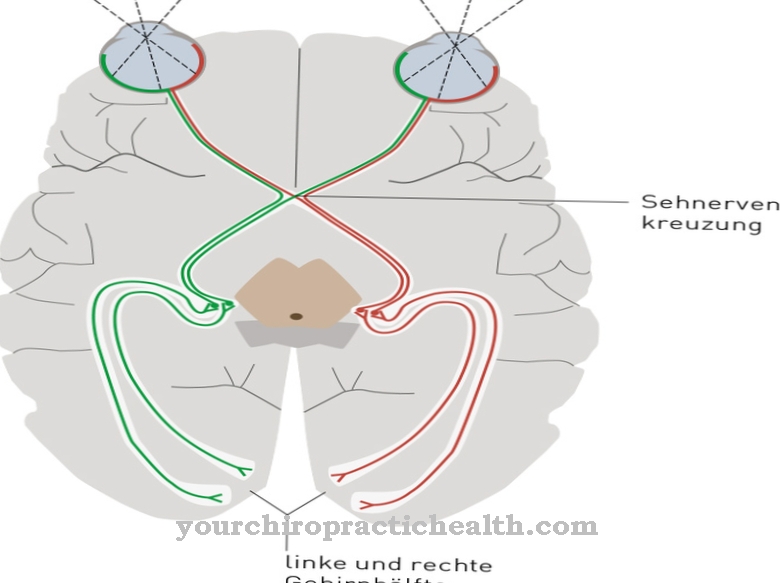
Τα ένζυμα που λείπουν λόγω γενετικών ελαττωμάτων αντισταθμίζονται με κανονικές ενδοφλέβιες εγχύσεις. Επειδή τα εγχυόμενα συνθετικά ένζυμα δεν μπορούν να διασχίσουν το φράγμα αίματος-εγκεφάλου λόγω του μοριακού τους μεγέθους, η θεραπεία λειτουργεί μόνο για ασθένειες αποθήκευσης λυσοσωμάτων που δεν επηρεάζουν το κεντρικό νευρικό σύστημα.
Τι είναι η θεραπεία αντικατάστασης ενζύμων;

Τα λυσοσώματα είναι ειδικά κυτταρικά οργανίδια στα οποία ξένες και ενδογενείς ουσίες διασπώνται και ανακυκλώνονται μερικώς. Απαιτούνται ειδικά ένζυμα υδρολύσεως για την αποικοδόμηση και μεταφορά των ουσιών. Αυτές είναι πρωτεάσες, νουκλεάσες, λιπάσες και ουσίες μεταφοράς.
Ένας αριθμός γνωστών γενετικών ελαττωμάτων μπορεί να οδηγήσει σε αποτυχία ορισμένων ενζύμων, έτσι ώστε ορισμένα προϊόντα αποικοδόμησης να συσσωρεύονται στα λυσοσώματα σε παθολογικές ποσότητες και να συσσωρεύονται έως ότου φθάσουν στην εξωκυτταρική μήτρα, δηλαδή στους διακυτταρικούς χώρους, με ανεξέλεγκτο τρόπο. Όλα τα γενετικά ελαττώματα που οδηγούν στην αποτυχία τουλάχιστον μιας απαραίτητης υδρολάσης συνοψίζονται με τον όρο λυσοσωμική ασθένεια αποθήκευσης. Η θεραπεία αντικατάστασης ενζύμων (ΕΡΤ, θεραπεία αντικατάστασης ενζύμωνχρησιμοποιείται για την αντικατάσταση των ενδογενών ενζύμων που λείπουν με συνθετικά παραγόμενα ένζυμα.
Επειδή οι υδρολάσες αποτελούνται από σχετικά μεγάλα μόρια, δεν μπορούν να απορροφηθούν από το έντερο χωρίς πρώτα να διαλυθούν και να απενεργοποιηθούν, έτσι ώστε να μπορούν να χορηγηθούν μόνο μέσω ενδοφλέβιας έγχυσης. Ωστόσο, το μέγεθος των ενζύμων μορίων αποτρέπει επίσης τη διέλευση του φραγμού αίματος-εγκεφάλου, έτσι ώστε η θεραπεία να μπορεί να είναι αποτελεσματική μόνο για ασθένειες αποθήκευσης λυσοσωμάτων που δεν επηρεάζουν το κεντρικό νευρικό σύστημα (ΚΝΣ).
Λειτουργία, αποτέλεσμα και στόχοι
Πάνω από 50 διαφορετικές λυσοσωμικές μεταβολικές διαταραχές είναι γνωστές, καθεμία από τις οποίες μπορεί να εντοπιστεί σε ένα μονογενετικό ελάττωμα. Οι ασθένειες αποθήκευσης λυσοσωμάτων μπορούν να χωριστούν σε επτά διαφορετικές κατηγορίες ανάλογα με τις υπερβολικά αποθηκευμένες ουσίες λόγω του υπάρχοντος ενζύμου ελαττώματος.
Οι βλεννοπολυσακχαρίτες και οι ολιγοσακχαρίτες είναι κατά κύριο λόγο κατάλληλες για ERT. Ο στόχος της ΕΡΤ είναι πάντοτε να αντισταθμίζει την ειδική ανεπάρκεια ενζύμων μέσω των τεχνητά παρεχόμενων ενζύμων προκειμένου να σταματήσει η ασθένεια ή τουλάχιστον μια ηπιότερη πορεία. Αναλυτικά, υπάρχουν διαθέσιμα ένζυμα αντικατάστασης για τις ακόλουθες λυσοσωμικές ασθένειες αποθήκευσης:
- Η νόσος του Gaucher
- Νόσος της Πομπηίας
- Νόσος Fabry
- Σύνδρομο Hurler-Pfaundler (βλεννοπολυσακχαρίτιδα I)
- Η νόσος του κυνηγού (βλεννοπολυσακχαρίτιδα II)
• Σύνδρομο Maroteaux-Lamy (βλεννοπολυσακχαρίτιδα VI) • Niemann-Pick B
Η νόσος του Gaucher είναι η πιο κοινή ασθένεια αποθήκευσης λυσοσωμάτων. Εμφανίζεται σε τρεις διαφορετικές παραλλαγές, δύο από τις οποίες επηρεάζουν επίσης το νευρικό σύστημα. Στη μη νευροπαθητική μορφή, ο σπλήνας επηρεάζεται ιδιαίτερα, ο οποίος διευρύνεται σε μεγάλο βαθμό και οδηγεί σε δευτερογενείς βλάβες όπως αναιμία και βλάβη στον μυελό των οστών. Τυπικά συμπτώματα είναι πόνος στα οστά και στις αρθρώσεις και διαταραχές του κυκλοφορικού. Η οξεία νευροπαθητική παραλλαγή της νόσου δείχνει μια σοβαρή πορεία και προσφέρει ελάχιστες πιθανότητες επιβίωσης πέρα από τα δύο πρώτα χρόνια της ζωής.
Η ασθένεια αποθήκευσης Η νόσος Pompe οφείλεται σε ανεπάρκεια του ενζύμου άλφα-1,4-γλυκοσιδάσης, το οποίο εμπλέκεται σε μεγάλο αριθμό μεταβολικών διεργασιών. Η νόσος του Pompe οδηγεί σε τεράστια διεύρυνση της καρδιάς (καρδιομεγαλία) και καρδιακή ανεπάρκεια. Υπάρχουν πρώιμα, σοβαρά μαθήματα που εμφανίζονται στους πρώτους μήνες της ζωής, καθώς και ήπιες μορφές που εμφανίζονται μόνο στα επόμενα χρόνια της ζωής.
Η ασθένεια Fabry προκαλείται από ένα γενετικό ελάττωμα που συνδέεται με το Χ, επομένως μόνο τα αγόρια και οι άνδρες μπορούν να επηρεαστούν από την ασθένεια αποθήκευσης. Η ασθένεια συνήθως οδηγεί σε συμπτώματα στην προχωρημένη παιδική ηλικία, όπως επεισόδια πόνου, κερατόματα του δέρματος, νεφρικά προβλήματα και καρδιακή βλάβη των μυών. Η ανεπάρκεια του ενζύμου άλφα-γαλακτοσιδάση Α οδηγεί σε συσσώρευση τριαεξωσίδης κεραμιδίου, η οποία είναι η αιτία της ενεργοποίησης των συμπτωμάτων, η οποία μπορεί επίσης να επηρεάσει το αυτόνομο νευρικό σύστημα.
Δεν είναι ασυνήθιστο να προκαλείται βλάβη σε καρδιακή προσβολή, έμφραγμα στα νεφρά ή ακόμη και εγκεφαλικό. Το σύνδρομο Hurler-Pfaundler είναι επίσης γνωστό ως βλεννοπολυσακχαρίτιδα, τύπου Ι και προκαλείται από διακοπή του μεταβολισμού της γλυκοζαμινογλυκάνης. Η ασθένεια σχετίζεται με μια μεγάλη ποικιλία συμπτωμάτων, όπως σοβαρή ψυχική δυσλειτουργία και σοβαρές σκελετικές αλλαγές. Η πορεία της νόσου είναι σοβαρή, έτσι ώστε το μέσο προσδόκιμο ζωής να είναι 11 έως 14 ετών. Η νόσος του Hunter αντιστοιχεί σε βλεννοπολυσακχαρίτιδα, τύπου 2 και - όπως η νόσος του Hurler - προκαλείται από ένα ελάττωμα που συνδέεται με το Χ. Η ασθένεια χαρακτηρίζεται από μαθήματα ποικίλης σοβαρότητας, από την εμφάνιση στην πρώιμη παιδική ηλικία έως ήπια μαθήματα που εμφανίζονται μόνο σε ενήλικες άνδρες.
Λόγω των πιο κοινών καρδιακών συμπτωμάτων όπως ελαττωμάτων καρδιακής βαλβίδας και προβλημάτων καρδιακών μυών, το προσδόκιμο ζωής κυμαίνεται από φυσιολογικό έως ελαφρώς περιορισμένο. Το σύνδρομο Maroteaux-Lamy (MPS VI) είναι μία από τις βλεννοπολυσακχαρίτες που κληρονομούνται με αυτοσωμικό υπολειπόμενο τρόπο επειδή το αιτιολογικό ελάττωμα του γονιδίου δεν βρίσκεται στο χρωμόσωμα Χ. Η ασθένεια είναι πολύ σπάνια, με μία περίπτωση ανά 455.000 γεννήσεις. Υπάρχουν γνωστές ήπιες και σοβαρές μορφές.
Τα συμπτώματα είναι διευρυμένο ήπαρ και σπλήνα, σύνδρομο καρπιαίου σωλήνα και αλλαγές στις καρδιακές βαλβίδες. Το Niemann-Pick B είναι μια λιπιδίωση σφιγγομυελίνης, η οποία είναι μία από τις λυσοσωμικές ασθένειες αποθήκευσης και προκαλείται από γενετικό ελάττωμα στο χρωμόσωμα 11. Ενώ ο τύπος Β της νόσου επηρεάζει κυρίως το ήπαρ και τον σπλήνα, ο τύπος Α έχει επίσης σημαντικά νευρωνικά προβλήματα.
Μπορείτε να βρείτε το φάρμακό σας εδώ
➔ Φάρμακα για τον πόνοΚίνδυνοι, παρενέργειες & κίνδυνοι
Δεδομένου ότι πολλές από τις ασθένειες αποθήκευσης λυσοσωμάτων που μπορούν να αντιμετωπιστούν με θεραπεία αντικατάστασης ενζύμων ακολουθούν σοβαρή πορεία με αντίστοιχα αυξημένο ποσοστό θνησιμότητας, ο μεγαλύτερος κίνδυνος στην ΕΡΤ είναι ότι το επιλεγμένο ένζυμο αντικατάστασης δεν λειτουργεί ή λειτουργεί μόνο πολύ ασθενώς.
Ένας άλλος κίνδυνος έγκειται λιγότερο στην ίδια τη θεραπεία παρά στο γεγονός ότι η υποκείμενη ασθένεια αναγνωρίζεται πολύ αργά, έτσι ώστε η ΕΡΤ να μπορεί να σταματήσει κατά τη διάρκεια της πορείας, αλλά η ζημιά που έχει ήδη προκληθεί δεν μπορεί να υποχωρήσει. Σχεδόν κάθε δευτερόλεπτο, ο ασθενής που λαμβάνει θεραπεία αντιδρά προσωρινά στις εγχύσεις με συμπτώματα όπως πυρετό και ρίγη. Οι λόγοι για αυτό δεν είναι ακόμη πλήρως κατανοητοί. Μερικοί ασθενείς αντιδρούν σχηματίζοντας αντισώματα και υπήρξαν γνωστές περιπτώσεις όπου οι ασθενείς αντέδρασαν με εξανθήματα και βρογχόσπασμο.



.jpg)



.jpg)
.jpg)
.jpg)
.jpg)