Υπάρχουν 45 διαφορετικά συνολικά λυσοσωμικές ασθένειες αποθήκευσης, που είναι μια ετερογενής ομάδα συγγενών μεταβολικών παθήσεων. Τα άτομα που πάσχουν από οποιαδήποτε από αυτές τις ασθένειες έχουν γενετικό ελάττωμα. Όλες οι ασθένειες αποθήκευσης έχουν ένα κοινό σημείο: ένα συγκεκριμένο ένζυμο είτε απουσιάζει είτε μόνο εν μέρει λειτουργικό.
Τι είναι η ασθένεια αποθήκευσης λυσοσωμάτων;

© designua - stock.adobe.com
Αυτές οι συγγενείς ασθένειες αποθήκευσης είναι σπάνιες, καθώς επηρεάζονται λιγότερα από πέντε στα 10.000 άτομα. Οι διάφορες ασθένειες έχουν πολύ διαφορετική πορεία και τα συμπτώματα μπορεί να ποικίλλουν ευρέως.
Οι πιο διάσημες μορφές του λυσοσωμική ασθένεια αποθήκευσης είναι η νόσος του Fabry, η νόσος του Gaucher, η νόσος Pompe και η βλεννοπολυσακχαρίτιδα (MPS). Συχνά αναφέρονται ως «ορφανά ιατρικής» επειδή η πορεία προς μια συγκεκριμένη διάγνωση και κατάλληλη θεραπεία μπορεί να είναι πολύ μεγάλη. Μερικές φορές μπορεί να χρειαστούν χρόνια για όσους επηρεάζονται για να μάθουν τι τους συμβαίνει.
αιτίες
Οι ασθένειες αποθήκευσης λυσοσωμάτων χαρακτηρίζονται από ορισμένες μορφές κληρονομικών μεταβολικών ασθενειών. Ο ασθενής δεν έχει ένα σημαντικό ένζυμο που διασφαλίζει ότι η μεταβολική ισορροπία λειτουργεί ομαλά. Στην λιγότερο έντονη μορφή, αυτό το ένζυμο τουλάχιστον δεν υπάρχει σε επαρκείς ποσότητες.
Το καθήκον των ενζύμων είναι η απόρριψη ρύπων και αποβλήτων που συσσωρεύονται στον ανθρώπινο οργανισμό μέσω του μεταβολισμού μέσω των λυσοσωμάτων, ή η επεξεργασία τους ξανά με τέτοιο τρόπο ώστε να μην εμφανίζονται συμπτώματα.
Εάν υπάρχει ανεπάρκεια ενζύμου, αυτός ο ομαλά λειτουργικός κύκλος απόρριψης δεν είναι πλέον εγγυημένος. Οι επιβλαβείς ουσίες εγκαθίστανται στα κύτταρα και διαταράσσουν τον μεταβολικό κύκλο. Στην αρχική φάση, οι διαταραχές δεν έχουν αισθητή επίδραση, υπάρχουν μόνο μερικοί περιορισμοί. Ωστόσο, εάν αυτή η μεταβολική διαταραχή παραμείνει χωρίς θεραπεία ως αποτέλεσμα ανεπάρκειας ενζύμου, τα συμπτώματα πολλαπλασιάζονται επειδή τα κύτταρα διευρύνονται πολύ.
Συμπτώματα, ασθένειες και σημεία
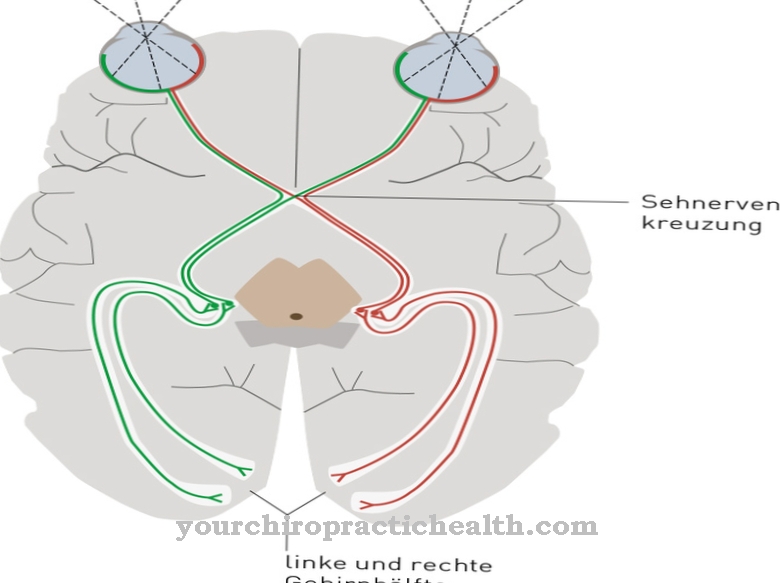
Στη χειρότερη περίπτωση, αυτά πηγαίνουν κάτω. Οι συνέπειες είναι η βλάβη στα οστά, το νευρικό σύστημα, ο σπλήνας, τα νεφρά, οι μύες ή η καρδιά. Λόγω της μειωμένης ή απουσίας ενζυματικής δραστηριότητας, η ασθένεια Fabry προκαλεί την αποθήκευση λίπους (σφαιροτριαζυλοκεραμίδη, Gb3) στα κύτταρα. Αυτές οι ανεπιθύμητες εναποθέσεις μπορεί να οδηγήσουν σε σοβαρό πόνο στα δάχτυλα των δακτύλων ή στα δάχτυλα, εγκεφαλικό επεισόδιο και νεφρική βλάβη.
Διάγνωση & πορεία της νόσου
Αυτή η κλινική εικόνα επηρεάζει ταυτόχρονα διαφορετικά συστήματα: αιμοφόρα αγγεία, νεφρά, καρδιά και νευρικό σύστημα. Η αυτοσωματική υπολειπόμενη νόσος Gaucher προκαλεί μετάλλαξη του ενζύμου "βήτα-γλυκοκερεοβροσιδάση" και οδηγεί σε συσσώρευση υποστρώματος εντός των κυττάρων, ειδικά στους μακροφάγους (φαγοκύτταρα) που ανήκουν στο δικτυο-ενδοθηλιακό σύστημα. Ο αριθμός αίματος αλλάζει, το ήπαρ και ο σπλήνας διογκώνονται και τα οστά πονάνε.
Η ασθένεια είναι προοδευτική και είναι ως επί το πλείστον εθνοτική, καθώς εμφανίζεται στις περισσότερες περιπτώσεις σε άτομα εβραϊκής καταγωγής. Η νόσος Pompe είναι επίσης γνωστή ως «ανεπάρκεια όξινης μαλτάσης». Η κλινική εικόνα ανήκει στην ομάδα της γλυκογένεσης τύπου ΙΙ. Τα προσβεβλημένα άτομα στερούνται του ενζύμου «άλφα-1,4-γλυκοσιδάση» (όξινη μαλτάση) ή δεν διατίθεται σε επαρκείς ποσότητες. Λόγω της εξασθενημένης διάσπασης του γλυκογόνου στους μύες, οι ασθενείς υποφέρουν από την καταστροφή των μυϊκών κυττάρων με τη μορφή αποθήκευσης σακχάρου.
Η βλεννοπολυσακχαρίτιδα τύπου Ι (MPS), επίσης γνωστή ως νόσος του Hunter, έχει διάφορες κλινικές αιτίες. Η νόσος του Hurler είναι η πιο σοβαρή μορφή και η νόσος του Scheie βρίσκεται στο τέλος της κλινικής παθογένεσης. Υπάρχουν μεταβάσεις διαφορετικών χαρακτηριστικών μεταξύ αυτών των δύο μορφών εξέλιξης. Η πιο εντυπωσιακή ιδιότητα είναι η εξασθενημένη διάσπαση των υδατανθράκων που συσσωρεύονται στα λυσοσώματα των κυττάρων.
Οι ασθενείς με ασθένεια κυνηγιού μπορεί να παρουσιάσουν βραχύ ανάστημα, διογκωμένη σπλήνα και ήπαρ, ακαθάριστα χαρακτηριστικά, πυκνό δέρμα, διογκωμένη γλώσσα και δυσκολία στην αναπνοή. Επιπλέον, ο σκελετός αλλάζει συχνά στην περιοχή της λεκάνης, της σπονδυλικής στήλης, των οστών χεριών και του κρανίου. Οι ομφαλικές και [[βουβωνικές κήλες] είναι δυνατές.
Επιπλοκές
Στις περισσότερες περιπτώσεις, τα συμπτώματα ή οι επιπλοκές εμφανίζονται πολύ αργά σε αυτήν την ασθένεια. Εξαιτίας αυτού, διαγιγνώσκεται αργά, καθιστώντας αδύνατη την έγκαιρη θεραπεία στις περισσότερες περιπτώσεις. Χωρίς θεραπεία, εμφανίζονται διάφορα παράπονα και βλάβες στα εσωτερικά όργανα καθώς εξελίσσεται η ασθένεια.
Τα νεφρά, το ήπαρ και ο σπλήνας επηρεάζονται ιδιαίτερα. Η καρδιά μπορεί επίσης να επηρεαστεί από αυτήν την ασθένεια, η οποία μπορεί να οδηγήσει σε καρδιακό θάνατο στη χειρότερη περίπτωση. Επιπλέον, συμβαίνει βλάβη στα νεφρά και εκείνοι που πάσχουν συχνά υποφέρουν από πόνο στα δάκτυλα των ποδιών ή των δακτύλων. Η παράλυση μπορεί επίσης να συμβεί εάν ο εγκέφαλος έχει υποστεί βλάβη από αυτήν την ασθένεια. Το ήπαρ και ο σπλήνας μπορούν να διογκωθούν και να προκαλέσουν σοβαρό πόνο επίσης.
Δεν είναι ασυνήθιστο τα οστά του πάσχοντος να είναι εύθραυστα και επίσης επώδυνα. Η θεραπεία αυτής της ασθένειας αποδεικνύεται δύσκολη. Σε πολλές περιπτώσεις, το προσδόκιμο ζωής του θιγόμενου ατόμου μειώνεται σημαντικά. Συνήθως δεν υπάρχουν ιδιαίτερες επιπλοκές κατά τη χρήση φαρμάκων. Ωστόσο, η θετική πορεία της νόσου δεν μπορεί να διασφαλιστεί σε κάθε περίπτωση.
Μπορείτε να βρείτε το φάρμακό σας εδώ
➔ Φάρμακα για τον πόνοΠότε πρέπει να πάτε στο γιατρό;
Η απώλεια μαλλιών, τα προβλήματα των αρθρώσεων και οι διαταραχές των οργάνων είναι πιθανά σημάδια ασθένειας αποθήκευσης λυσοσωμάτων Συνιστάται επίσκεψη στο γιατρό εάν τα συμπτώματα συνεχίζουν να επαναλαμβάνονται ή εμφανίζονται ξαφνικά χωρίς να εντοπιστεί αιτία. Εάν τα συμπτώματα σχετίζονται με προηγουμένως διαγνωσμένο ελάττωμα ενζύμου ή άλλη σοβαρή ασθένεια, θα πρέπει να συμβουλευτείτε τον υπεύθυνο γιατρό. Μια ασθένεια αποθήκευσης χωρίς θεραπεία μπορεί να οδηγήσει σε άνοια, στειρότητα, νευροπάθειες και άλλες επιπλοκές, μερικές από τις οποίες είναι απειλητικές για τη ζωή. Επομένως, πρέπει να εξεταστούν όλα τα πιθανά συμπτώματα, ακόμη και αν δεν υπάρχει συγκεκριμένη υποψία.
Τα συμπτώματα της ασθένειας αποθήκευσης λυσοσωμάτων μπορεί να εμφανιστούν σε φάσεις ή να αναπτυχθούν ύπουλα, αλλά πάντοτε απαιτούν εξέταση και θεραπεία. Τα επηρεαζόμενα άτομα είναι καλύτερα να μιλήσουν απευθείας στον οικογενειακό γιατρό τους ή σε έναν παθολόγο. Η πραγματική θεραπεία πραγματοποιείται συνήθως σε εξειδικευμένη κλινική για εσωτερικές ασθένειες, αν και η φυσιοθεραπεία ή η ψυχοθεραπεία μπορούν να συνδεθούν ανάλογα με τα συμπτώματα. Συγκεκριμένα, τα θεραπευτικά μέτρα υποδεικνύονται λόγω της συχνά αρνητικής πορείας της νόσου.
Θεραπεία & Θεραπεία
Ανάλογα με το πόσο νωρίς γίνεται μια επαρκής διάγνωση, αυτές οι κληρονομικές ασθένειες μπορούν να αντιμετωπιστούν πολύ καλά με θεραπεία αντικατάστασης ενζύμου, έτσι ώστε τα άτομα που έχουν προσβληθεί να έχουν πολύ λιγότερα συμπτώματα και, επομένως, καλύτερη ποιότητα ζωής. Αυτή η θεραπεία αντικατάστασης χρησιμοποιείται σύμφωνα με την κλινική εικόνα.
Τα άτομα που πάσχουν από τη νόσο του Gaucher δεν διαθέτουν το «ένζυμο ß-γλυκοκερεβροσιδάση», το οποίο παράγεται βιοτεχνολογικά και εγχύεται στο σώμα του ασθενούς. Τα λυσοσώματα δρουν αποτελεσματικά και είναι ικανά να απορροφούν ουσίες από το άμεσο περιβάλλον τους. Για το λόγο αυτό, τα τεχνητά χρησιμοποιούμενα ένζυμα τροποποιούνται με τέτοιο τρόπο ώστε να μπορούν να παρέχονται στα λυσοσώματα με ιδανικό τρόπο.
Οι μακροφάγοι (φαγοκύτταρα) διασπώνουν τις γλυκοκερεοβροσίδες που έχουν συσσωρευτεί στα κύτταρα. Αυτή η θεραπεία μπορεί να συγκριθεί με τη θεραπεία με ινσουλίνη για σακχαρώδη διαβήτη, με τη διαφορά ότι δεν είναι ορμόνη που λείπει αλλά ένζυμο που δεν παρέχεται. Το σώμα διασπά τακτικά όλες τις ουσίες, συμπεριλαμβανομένου του παρεχόμενου τεχνητού ενζύμου.
Λόγω αυτής της τακτικής κατανομής της ουσίας, οι ασθενείς πρέπει να υποβάλλονται τακτικά σε αυτήν τη θεραπεία έγχυσης μέχρι το τέλος της ζωής τους. Η θεραπεία αντικατάστασης ενζύμων δεν δρα συμπτωματικά, αλλά καταπολεμά άμεσα την αιτία της κληρονομικής νόσου. Οι γιατροί αποκαλούν αυτή τη θεραπεία αιτιώδη. Οι αρχές της θεραπείας πρέπει να χρησιμοποιούνται και για τις τέσσερις από τις προαναφερθείσες κοινές ασθένειες αποθήκευσης.
Οι ασθενείς με Pompe λαμβάνουν επίσης θεραπεία με έγχυση. Σε αυτήν την ασθένεια, το ανύπαρκτο ένζυμο "acid alfa glukosidase" παρέχεται και βοηθά στη διάσπαση του γλυκογόνου που έχει συσσωρευτεί στα λυσοσώματα των μυών. Σε ασθενείς με τύπο νόσου «βλεννοπολυσακχαρίτιδα τύπου Ι», το λυσοσωμικό ένζυμο «άλφα-iduronidase» δεν υπάρχει ή δεν υπάρχει σε επαρκείς ποσότητες. Είναι μια από τις πιο σπάνιες ασθένειες αποθήκευσης στις οποίες τα μόρια σακχάρου συσσωρεύονται σε όργανα και ιστούς.
Εάν η διαδικασία είναι φυσιολογική, το ένζυμο διασπά τους βλεννοπολυσακχαρίτες. Τα μόρια σακχάρου είναι αλυσοπρίονα μακράς διάρκειας και εμπλέκονται στην ανάπτυξη υποστηρικτικού και συνδετικού ιστού, για παράδειγμα οστών, δέρματος, υγρών αρθρώσεων και χόνδρου. Εάν η φυσιολογική πορεία της αποικοδόμησης διαταραχθεί λόγω της έλλειψης ενζύμου, οι παθολογικές γλυκοζαμινογλυκάνες (GAG) συσσωρεύονται στα μεμονωμένα κύτταρα. Οι μελλοντικές επιλογές θεραπείας στοχεύουν στη λήψη δισκίων.
Προοπτικές και προβλέψεις
Η πρόγνωση για την ασθένεια αποθήκευσης είναι κακή. Μια γενετική διάθεση βρέθηκε να είναι η αιτία της διαταραχής της υγείας. Οι νομικές απαιτήσεις απαγορεύουν στους ιατρούς και τους επιστήμονες να αλλάζουν την ανθρώπινη γενετική. Για το λόγο αυτό, η ασθένεια παραμένει ισόβια και δεν έχει προοπτική ανάρρωσης.
Ο θεράπων ιατρός επικεντρώνεται στη θεραπεία των συμπτωμάτων που προκύπτουν. Εάν αφεθεί χωρίς θεραπεία, διάφορα παράπονα θα αυξάνονται με την πάροδο του χρόνου. Το οστικό σύστημα είναι κατεστραμμένο και προκύπτουν προβλήματα των οργάνων. Στη χειρότερη περίπτωση, τα εσωτερικά όργανα θα δυσλειτουργούν και τελικά η λειτουργία τους θα αποτύχει. Αυτό απειλεί τον ενδιαφερόμενο με πρόωρο θάνατο.
Η πρόκληση της νόσου έγκειται στη διάγνωση. Σε μεγάλο αριθμό ασθενών, αξιοσημείωτα και έντονα αντιληπτά παράπονα εμφανίζονται μόνο αργότερα στη ζωή τους. Ως αποτέλεσμα, η γενετική διαταραχή παραμένει απαρατήρητη για μεγάλο χρονικό διάστημα και η έγκαιρη θεραπεία της νόσου είναι δύσκολη. Όσο αργότερα γίνει διάγνωση, τόσο πιο δυσμενής είναι η περαιτέρω πορεία. Σε προχωρημένο στάδιο της νόσου, τα εσωτερικά όργανα ή οι αρθρώσεις έχουν ήδη υποστεί σοβαρή βλάβη. Απαιτούνται χειρουργικές επεμβάσεις και εάν η ασθένεια εξελίσσεται δυσμενώς, μόνο ένα όργανο δότη μπορεί να σώσει τη ζωή του ατόμου που πάσχει. Η έγκαιρη θεραπεία είναι επομένως απαραίτητη για μια βελτιωμένη πρόγνωση.
πρόληψη
Δεδομένου ότι είναι ένα συγγενές γενετικό ελάττωμα που εμποδίζει την έκφραση ενός ενζύμου, αυτή η ασθένεια δεν μπορεί να αντιμετωπιστεί προληπτικά. Ωστόσο, τα τελευταία επιτεύγματα γενετικής μηχανικής θα μπορούσαν να προσφέρουν μια προσέγγιση σε αυτόν τον τομέα.
Μετέπειτα φροντίδα
Με αυτή την ασθένεια, οι άνθρωποι πάσχουν από διάφορες επιπλοκές και ασθένειες. Κατά κανόνα, όλα αυτά έχουν πολύ αρνητική επίδραση στην ποιότητα ζωής του θιγόμενου ατόμου, οπότε η διάγνωση πρέπει να γίνει πολύ νωρίς. Όσο νωρίτερα ζητηθεί η γνώμη ενός γιατρού, τόσο καλύτερη είναι συνήθως η περαιτέρω πορεία αυτής της νόσου.
Η σοβαρότητα αυτής της ασθένειας μπορεί να ποικίλλει σε μεγάλο βαθμό, έτσι ώστε μια γενική πρόβλεψη συχνά δεν είναι δυνατή. Όσοι πάσχουν από σοβαρή βλάβη στα εσωτερικά όργανα. Τα νεφρά και η καρδιά επηρεάζονται κυρίως, έτσι ώστε το παιδί να μπορεί να πεθάνει τις πρώτες ημέρες, εάν τα συμπτώματα δεν διορθωθούν εγκαίρως. Υπάρχουν επίσης εναποθέσεις λίπους σε διάφορα μέρη του σώματος.
Τα δάχτυλα και τα δάχτυλα των ποδιών επηρεάζονται ιδιαίτερα, γεγονός που μπορεί να οδηγήσει σε σημαντικά μειωμένη αισθητική για το άτομο που πάσχει. Κατά κανόνα, η βλάβη στα νεφρά και στον εγκέφαλο συμβαίνει στο επόμενο στάδιο, έτσι ώστε το άτομο που επηρεάζεται να πεθάνει ως αποτέλεσμα αυτής της βλάβης. Οι γονείς και οι συγγενείς συχνά πάσχουν από κατάθλιψη ή άλλες ψυχικές διαταραχές λόγω της ασθένειας.
Μπορείτε να το κάνετε μόνοι σας
Οι ασθένειες αποθήκευσης λυσοσωμάτων συχνά απαιτούν εντατική ιατρική περίθαλψη. Συχνά δεν υπάρχουν αρκετές ευκαιρίες για αυτοβοήθεια. Οι γονείς των παιδιών που πλήττονται συχνά αντιμετωπίζουν έντονο άγχος στο περιβάλλον του σπιτιού τους επειδή το παιδί τους χρειάζεται συνεχή φροντίδα και προσοχή.
Οι κλινικές εικόνες των μεμονωμένων ασθενειών αποθήκευσης είναι διαφορετικές. Υπάρχουν τόσο εύκολες όσο και πολύ δύσκολες μορφές. Ένα παράδειγμα είναι η νόσος του Gaucher. Η βοήθεια των γονέων περιορίζεται συχνά στη σίτιση του παιδιού με σοβαρή αναπηρία. Σε ήπιες περιπτώσεις, το προσδόκιμο ζωής μπορεί να είναι σχεδόν φυσιολογικό. Ωστόσο, απαιτείται συνεχής ιατρική παρακολούθηση για την αποφυγή πιθανών επιπλοκών. Η τακτική σωματική δραστηριότητα είναι μία από τις συνοδευτικές θεραπείες που μπορούν επίσης να πραγματοποιηθούν στο σπίτι. Επιπλέον, πρέπει να γίνει μια ενδελεχής εξέταση διαλογής καρκίνου. Αυτό απαιτεί συνεχείς επισκέψεις στο γιατρό με το παιδί τους από τους γονείς. Το ίδιο ισχύει και για άλλες ασθένειες αποθήκευσης λυσοσωμάτων.
Στην περίπτωση ορισμένων ασθενειών, εκτός από τις σωματικές αναπηρίες, μπορεί επίσης να εμφανιστούν ψυχικές διαταραχές, οι οποίες εξακολουθούν να απαιτούν ειδική υποστήριξη. Σε ήπιες μορφές ορισμένων ασθενειών, όπως η νόσος του Hunter, αρχικά εμφανίζονται μόνο σκελετικές αλλαγές και δυσμορφισμός του προσώπου. Εδώ, ωστόσο, ο πάσχων ασθενής είναι συχνά σε θέση να ζήσει μια ανεξάρτητη ζωή. Ωστόσο, απαιτούνται επίσης συνεχείς ιατρικές εξετάσεις για να αποκλειστούν πιθανές επιπλοκές όπως καρδιακή ανεπάρκεια ή αναπνευστικές παθήσεις. Ο ασθενής μπορεί να αντιμετωπίσει ψυχολογικό άγχος που προκαλείται από σωματικές παραμορφώσεις μέσω ψυχολογικής συμβουλευτικής.



.jpg)



.jpg)
.jpg)
.jpg)
.jpg)